3D Chromatin Organization
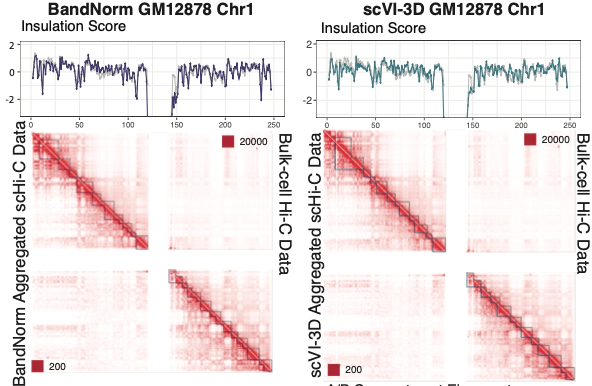 Photo by rawpixel on Unsplash
Photo by rawpixel on Unsplash
Project Introduction
The role that chromatin architecture plays in the mechanism of gene regulation has undergone a dramatic transformation with the emergence of Hi-C and other 3C-derived high throughput technology for interrogating the three-dimensional configurations of the genome and identifying regions that are in close spatial proximity in a genome-wide fashion. As the sequencing depth grows, the resolution to investigate the 3D chromatin interactions goes from Megabase scale to kilobase or even hundred-base genomic scale. Furthermore, with the development of other “C”-related techniques, for instance, HiChIP, DNaseHiC, Capture Hi-C, etc., we will be able to investigate all kinds of genomic interactions at ultra-high resolution across the whole genome from the anchor regions that can be associated with clinically relevant loci such as regulatory elements, single nucleotide polymorphisms (SNPs) from GWAS studies, chromatin domain boundaries or promoters. Analysis of 3D genome data will resolve many fundamental and long-standing questions involving the mechanism of gene regulation from distal non-coding regulatory variants, linking novel SNPs to putative disease-associated genes related to all sorts of disorders, the connection between chromatin domain structure variation and cancer or other genetic diseases.
Such technological advances in 3D genomics lead to great needs for new statistical models and computational tools to harness the embedding conformation information present in Hi-C data. To achieve decent interaction resolution, namely, how specific the region can be if we zoom in the genome to the largest extent, one study usually has billions of reads for each biological replicate. Hence, it poses a tough challenge on the model which should be theoretically sound and practically applicable and implementable within a short time. Realizing the demand in the field, I have developed a few fundamental methods, mHi-C, FreeHi-C, FreeHi-C SpikeIn, BandNorm and scVI-3D, with the corresponding software packages or pipelines to address many basic needs in dealing with Hi-C data.
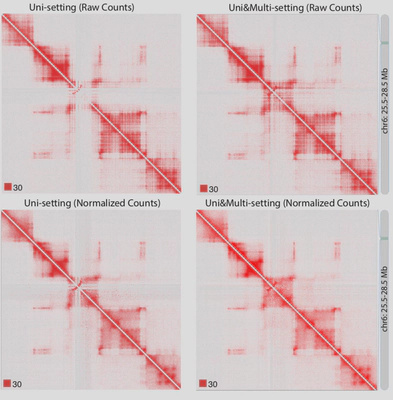
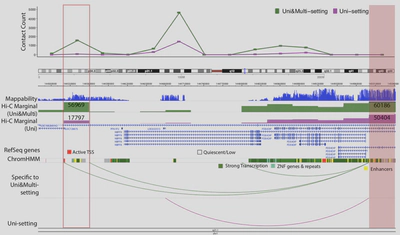
Due to the importance of sequencing depth in Hi-C analysis, I developed mHi-C (Zheng et al. 2019 eLife) to utilize multi-mapping reads that are discarded in Hi-C benchmark analysis pipeline. Multi-mapping reads are reads originated from repetitive regions of the genome hence they can be aligned to multiple positions. The ambiguity of multi-reads alignment renders them to be deleted wasting ~20% of the sequencing depth which can be translated into hundreds of millions of reads for one biological replicate. Through a hierarchical model that probabilistically allocates Hi-C multi-reads to their most likely genomic origins by utilizing specific characteristics of the paired-end reads of the Hi-C assay, the chromatin domain boundaries are refined and we can also detect novel promoter-enhancer interactions that involve disease-associated genes. Moreover, mHi-C, itself is programmed into a python pipeline with C acceleration, can process from raw data and output valid genomic interactions. Another urgent demand from both experimental labs and computational groups is a tool to fast simulate Hi-C contact matrices to work as biological replicates to improve the detection power or employed as a test of models under development. Also, the tool should be easy to use for researchers from all kinds of background. Therefore, I have developed FreeHiC (Zheng et al. 2020. Nature Methods) and FreeHi-C SpikeIn (Zheng et al. 2021. Methods) which empirically learns the interacting fragment behaviors, sequence characters, according to the Hi-C experimental protocol procedures. Sequently, with the user-controlled level of noise being inserted into the simulation process, a similar Hi-C interaction matrix is generated.
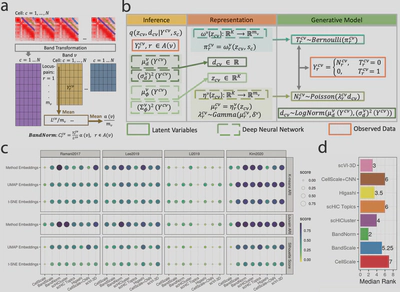
Recently, single-cell high-throughput chromatin conformation capture methodologies (scHi-C) enable profiling long-range genomic interactions. However, data from these technologies are prone to technical noise and biases that hinder downstream analysis. Collaborating with Keles Lab, we developed a normalization approach, BandNorm, and a deep generative modeling framework, scVI-3D, to account for scHi-C specific biases. In benchmarking experiments, BandNorm yields leading performances in a time and memory efficient manner for cell-type separation, identification of interacting loci, and recovery of cell-type relationships, while scVI-3D exhibits advantages for rare cell types and under high sparsity scenarios. Application of BandNorm coupled with gene-associating domain analysis reveals scRNA-seq validated sub-cell type identification.
Publication
-
Zheng Y+, Shen S+, Keleş S. Normalization and De-noising of Single-cell Hi-C Data with BandNorm and scVI-3D. Accepted by Genome Biology. 2022. (+: co-first authors)
-
Cheng J, Clayton J, Acemel R, Zheng Y, Taylor R, Keleş S, Harley J, Quail E, Gómez-Skarmeta J and Ulgiati D. Regulatory architecture of the RCA gene cluster captures an intragenic TAD boundary and enhancer elements in B cells. Frontiers in Immunology, section B Cell Biology. 2022.
-
Zheng Y, Zhou P, Keleş S. FreeHi-C Spike-in Simulations for Benchmarking Differential Chromatin Interaction Detection. Methods. 2021.
-
Huang K, Wu Y, Shin J, Zheng Y, Siahpirani A, Lin Y, Ni Z, Chen J, You J, Keleş S, Wang D, Roy S, Lu Q. Transcriptome-wide transmission disequilibrium analysis identifies novel risk genes for autism spectrum disorder. PLOS Genetics. 2021.
-
Zheng Y, Keleş S. FreeHi-C simulates high-fidelity Hi-C data for benchmarking and data augmentation. Nature Methods. 2020.
-
The ENCODE Project Consortium, et al. Expanded Encyclopedias of DNA Elements in the Human and Mouse Genomes. Nature. 2020 .
-
The ENCODE Project Consortium, Snyder, M.P., Gingeras, T.R., Moore, J.E., Weng, Z., Gerstein, M.B., Ren, B., Hardison, R.C., Stamatoyannopoulos, J.A., Graveley, B.R., Feingold, E.A. and Pazin, M.J. Perspectives on ENCODE. Nature. 2020.
-
Zheng Y, Ay F, Keleş S. Generative modeling of multi-mapping reads with mHi-C advances analysis of Hi-C studies. eLife. 2019.